
Manojkumar Kumaran
About Me
Hi! I'm Manojkumar Kumaran and Bioinformatician
I am a Computational Neurobiologist and Software Developer passionate about understanding the molecular and circuit mechanisms underlying neurological disorders and regeneration. I specialize in single-cell multi-omics, Hi-C, ChIP-seq, and ATAC-seq analysis, integrating computational and experimental approaches to study axon regeneration, neurodegeneration, and brain plasticity. My research focuses on how genomic architecture, transcription factors, and chromatin accessibility shape neuronal function in health and disease. Beyond research, I have extensive experience in software development, building automated bioinformatics pipelines, and applying machine learning for large-scale genomic analysis. I thrive on solving complex biological problems using computational tools and data-driven insights. In my free time, I enjoy wildlife photography, gaming, and cooking.
Birthday : 29 September 1991
Age : 33
Email : manojkumarbioinfo@gmail.com
Degree : PhD
Phone : +91 7708556754
City : Madurai
Freelance : Available
Nationality : Indian
Languages : English, Tamil
Current Role : Research Associate
Research Interests : Multi-Omics, AI in Neuroscience
Hobbies : Wildlife Photography, Gaming, Cooking
Shell, Python, R, Docker
Clinical WES/WGS Sequecning analysis
Single Cell Genomics sequecning analysis
Bulk ATAC, Chip, Cut&Run, HiC analysis
Structural bioinformatics
Machine Learning, Animal Behavior
Java, Html, Bootstrap
Server and Network Manager
Education
2015 - 2021
PhD In bioinformatics, at Arvind Medical Research Foundation
I developed an eye disease-specific variant prioritization tool, eyeVarP, which utilizes a random forest algorithm to analyze pathogenic variants associated with eye diseases and other disorders. Additionally, I created the VarP tool, a generalized pipeline designed to filter missense and insertion-deletion variants and predict their pathogenicity from exome or genome sequencing data.
2009 - 2015
Master of Technology (M.Tech) in Bioinformatics
I pursued a six-year integrated M.Tech program in Bioinformatics at Bharathidasan University, gaining expertise in computational biology, genomics, proteomics, and data-driven research methodologies. Throughout my academic journey, I developed a strong foundation in biological data analysis, machine learning applications in genomics, structural bioinformatics, and molecular modeling. I actively worked on research projects related to genome sequencing, variant analysis, and computational drug discovery, equipping myself with both wet-lab and computational skills essential for bioinformatics research.
Experience
2022 - Present
Research Associate, CSIR-CCMB
Developed an automated pipeline for analyzing single-cell RNA-seq, single-cell ATAC-seq, CUT&RUN, Hi-C, and bulk ATAC-seq data. Built a machine learning model for mouse behavioral analysis. Maintained the lab website and the GitHub repository.
2021 - 2022
Data Scientist, INDX Technologies
Developed an automated tool for analyzing Whole Exome Sequencing (WES) data from cancer patient datasets. Created an automated annotation tool to classify genetic variants based on available drug information.
2015 - 2021
Junior Research Fellow, Aravind Medical Research Foundation
Developed and benchmarked analytical tools and pipelines for Next-Generation Sequencing (NGS) data. Applied biostatistics and bioinformatics algorithms to integrate multi-level biological data with functional genomics for hypothesis generation, variant identification, and variant prioritization. Collaborated across multiple disciplines and contributed to various research projects. Maintained the lab website and the GitHub repository.
Publications
Total Citations: 91 | H-index: 4 | Research Score: 69.5 | Reads: 3840
PATZ1 Reinstates a Growth-Permissive Chromatin Landscape in Adult Corticospinal Neurons After Injury
Anisha S Menon, Manojkumar Kumaran et al. biorxiv (2025)
Show Abstract
Our research reveals how nerve cell growth genes become restricted during development through two distinct waves of chromatin closure. We found that injuries closer to the brain trigger broader chromatin hanges than distant injuries. Importantly, introducing the PATZ1 transcription factor after injury successfully reopened chromatin and reactivated developmental growth pathways. This work defines the timing of hromatin restriction in CNS neurons and demonstrates it can be reversed to potentially support regeneration.
Single-nuclei sequencing reveals a robust corticospinal response to nearby axotomy but overall insensitivity to spinal injury
Manojkumar Kumaran et al. Journal of Neuroscience (2025)
Show Abstract
The ability of neurons to sense and respond to damage is crucial for maintaining homeostasis and facilitating nervous system repair. For some cell types, notably dorsal root ganglia and retinal ganglion cells, extensive profiling has uncovered a significant transcriptional response to axon injury, which influences survival and regenerative outcomes. In contrast, the injury responses of most supraspinal cell types, which display limited regeneration after spinal damage, remain mostly unknown. In this study, we used single-nuclei sequencing in adult male and female mice to profile the transcriptional responses of diverse supraspinal cell types to spinal injury. Surprisingly, thoracic spinal injury induced only modest changes in gene expression across all populations, including corticospinal tract (CST) neurons. Additionally, CST neurons exhibited minimal response to cervical injury but showed a much stronger reaction to intracortical axotomy, with upregulation of numerous regeneration and apoptosis-related transcripts shared with injured DRG and RGC neurons. Thus, the muted response of CST neurons to spinal injury is linked to the injury's distal location, rather than intrinsic cellular characteristics. More broadly, these findings indicate that a central challenge for enhancing regeneration after a spinal injury is the limited detection of distant injuries and the subsequent modest baseline neuronal response.
Investigating druggable kinases for targeted therapy in cancer
Kumar Jeyaprakash, Manojkumar Kumaran et al. Journal of Human Genetics (2024)
Show Abstract
Retinoblastoma (RB) is a childhood retinal neoplasm and commonly treated with cytotoxic chemotherapeutic agents. However, these therapeutic approaches often lead to diverse adverse effects. A precise molecular therapy will alleviate these side effects and offer better treatment outcomes. Over the years, kinases have become potential drug targets in cancer therapy. Hence, we aimed to investigate genetic alterations of putative kinase drug targets in RB. Targeted exome sequencing was performed on 35 RB tumors with paired blood samples using a gene panel consisting of 29 FDA-approved kinase genes. Single nucleotide variants were analyzed for pathogenicity using an in-house pipeline and copy number variations (CNVs) were detected by a depth of coverage and CNVPanelizer. The correlation between genetic changes and clinicopathological features was assessed using GraphPad Prism. Three somatic mutations, two in ERBB4 and one in EGFR were identified. Two of these mutations (ERBB4 c.C3836A & EGFR c.A1196T) were not reported earlier. CNV analysis revealed recurrent gains of ALK, MAP2K2, SRC, STK11, and FGFR3 as well as frequent losses of ATM, PI3KCA and ERBB4. Notably, nonresponsive tumors had a higher incidence of amplifications in clinically actionable genes such as ALK. Moreover, ALK gain and ATM loss were strongly correlated with optic nerve head invasion. In conclusion, our study revealed genetic alterations of druggable kinases in RB, providing preliminary insights for the exploration of kinase-targeted therapy in RB.
Comprehensive ctDNA profiling reveals potential resistance mechanisms
Shafi, Gowhar , Manojkumar Kumaran et al. Cancer Research (2024)
Show Abstract
Background: Genomic profiling has revolutionized precision oncology impacting the diagnosis, prognosis, and therapy decisions. Considering high spatiotemporal diversity and heterogenicity of breast tumor-cell genomes, small-gene panels often fail to capture rare but important genomic alterations. Conversely, comprehensive ctDNA sequencing approaches enable the identification of under characterized 'long tailed driver' genomic alterations and capture Intra and inter metastatic heterogeneity. Here, we demonstrate the clinical utility of comprehensive genome profiling with higher sensitivity to predict the possibility of metastasis in early-stage breast cancer patients. Methods: We retrospectively analyzed ctDNA and genomic DNA (gDNA) from FFPE samples as well as circulating tumor cells (CTC) in 10 treatment-naive hormone positive and HER2 negative, primary-stage breast cancer patients [GS1] using the OncoIndx comprehensive 600 gene panel. The panel captures all important cancer-relevant genomic alterations including Tumor Mutation Burden (TMB), Micro Satellite Instability (MSI), homologous recombination deficiency (HRD) prediction, and cfDNA tumor fraction (TF). CTCs were enumerated from 1.5 ml of blood using the OncoDiscover platform approved by the Drug Controller General of India having anti-EpCAM antibody-mediated immunomagnetic nanoparticles. CTCs were confirmed for cytokeratin 18+ and DAPI + markers and the absence of CD45. Results: Comprehensive genomic profile obtained from ctDNA and gDNA from FFPE of early-stage breast cancer patients predominantly exhibited the presence of alterations in PIK3CA and ESR1 signaling pathways. PIK3CAmutations were present in 77% and 44% of baseline ctDNA and gDNA samples, while ESR1 mutations were present in 44% and 22% of baseline ctDNA and gDNA, respectively. In addition, we observed about 70% additional driver mutations in ctDNA samples suggesting shedding of ctDNA together with CTC (80% positive), a likely positive biomarker of metastasis. About 50% of the patients showed higher TMB and HRD. Notably, TF representing ctDNA varied between 13% to 27% in blood samples with a corresponding ploidy range of 2.9 to 4.7. Surprisingly, ~50% of the patient population matched the mutation profile of clinically confirmed metastatic patients. All the patients harboring potential metastatic driver alterations showed the presence of CTCs in peripheral blood. Conclusions: Comprehensive ctDNA genomic profiling showed potential metastasis driving alterations suggesting the role of ctDNA-based liquid biopsy to predict metastasis in early breast cancer patients. We observed enhanced TF at the time of diagnosis, possibly due to the presence of distant metastasis, high disease burden, and aggressive tumor biology. Our results suggest that ctDNA dynamics at the time of disease presentation can predict early metastasis, and may demonstrate the divergent response of tumor heterogeneity to treatment in early-stage breast cancer.
eyeVarP: a computational framework for the identification of pathogenic variants specific to eye disease
Manojkumar Kumaran et al. Genetics in Medicine (2023)
Show Abstract
Purpose: Disease-specific pathogenic variant prediction tools that differentiate pathogenic variants from benign have been improved through disease specificity recently. However, they have not been evaluated on disease-specific pathogenic variants compared with other diseases, which would help to prioritize disease-specific variants from several genes or novel genes. Thus, we hypothesize that features of pathogenic variants alone would provide a better model. Methods: We developed an eye disease–specific variant prioritization tool (eyeVarP), which applied the random forest algorithm to the data set of pathogenic variants of eye diseases and other diseases. We also developed the VarP tool and generalized pipeline to filter missense and insertion-deletion variants and predict their pathogenicity from exome or genome sequencing data, thus we provide a complete computational procedure. Results:eyeVarP outperformed pan disease–specific tools in identifying eye disease–specific pathogenic variants under the top 10. VarP outperformed 12 pathogenicity prediction tools with an accuracy of 95% in correctly identifying the pathogenicity of missense and insertion-deletion variants. The complete pipeline would help to develop disease-specific tools for other genetic disorders. Conclusion:eyeVarP performs better in identifying eye disease–specific pathogenic variants using pathogenic variant features and gene features. Implementing such complete computational procedure would significantly improve the clinical variant interpretation for specific diseases.
Clinical reassessments and whole-exome sequencing
Manojkumar Kumaran et al. Ophthalmic Genetics (2021)
Show Abstract
Background: The diagnosis of retinal dystrophies can be challenging due to the spectrum of protean phenotypic manifestations. This study employed trio-whole-exome sequencing (trio-WES) to unveil the genetic cause of an inherited retinal disorder in a south Indian family. Materials and methods: Proband's initial ophthalmic examinations was performed in the year 2016. WES was performed on a proband-parent trio to identify causative mutation followed by Sanger validation, segregation analysis, sequence and structure-based computational analysis to assess its pathogenicity. Based on the genetic findings, detailed clinical reassessments were performed in year 2020 for the proband and available family members. Results: WES revealed a novel homozygous BEST1 mutation c.G310A (p.D104N) in the proband and heterozygous for the parents, indicating autosomal recessive inheritance. Segregation analysis showed heterozygous mutation in maternal grandfather and normal genotype for younger brother and maternal grandmother. Moreover, the structure-based analysis revealed the mutation p.D104N in the cytoplasmic domain, causing structural hindrance by altering hydrogen bonds and destabilizing the BEST1 protein structure. Proband's clinical assessments were consistent with autosomal recessive bestrophinopathy (ARB) phenotype. Additionally, characteristic absent light rise and decreased light peak-to-dark trough ratio (LP:DT) was observed bilaterally in EOG. Conclusions: Our study demonstrates the utility of WES and clinical re-evaluations in establishing the precise diagnosis of autosomal recessive bestrophinopathy associated with a novel mutation, thus expanding the BEST1-related mutation spectrum.
Bioinformatics for Whole Exome Studies
Manojkumar Kumaran et al. Bioinformatics and Human Genomics Research (2021)
Show Abstract
Recent advances in next-generation sequencing (NGS) methods have greatly promoted the NGS-based genome/exome studies in discovering genetic variants for human diseases. Whole-genome sequencing (WGS) remains prohibitively expensive and requires concurrent development of bioinformatics approaches. Whole exome sequencing (WES), focusing on only the protein-coding sequence of the human genome, is now a widely used cost-effective approach that could identify disease-causing variants. Here, we provide the performance assessment of various aligners and variant calling tools using simulated and standard human exome datasets and highlight their combinations that improve the detection of single nucleotide variants (SNVs) and InDels. Further, we provide general recommendations for bioinformatic analysis for WES methods, focusing on variant filtering and prioritization for clinical use.
Whole-exome sequencing identifies multiple pathogenic variants
Manojkumar Kumaran et al. Indian Journal of Ophthalmology (2021)
Show Abstract
Purpose: To identify the pathogenic variants associated with primary open-angle glaucoma (POAG) using whole-exome sequencing (WES) data of a large South Indian family. Methods: We recruited a large five-generation South Indian family (n = 84) with a positive family history of POAG (n = 19). All study participants had a comprehensive ocular evaluation. We performed WES for 16 samples (nine POAG and seven unaffected controls) since Sanger sequencing of the POAG candidate genes (MYOC, OPTN, and TBK1) showed no genetic variation. We used an in-house pipeline for prioritizing the pathogenic variants based on their segregation among the POAG individual. Results: We identified one novel and five low-frequency pathogenic variants with consistent co-segregation in all affected individuals. The variant c.G3719A in RPGR-interacting domain of RPGRIP1 that segregated heterozygously with the six POAG cases is distinct from variants causing photoreceptor dystrophies, reported affecting the RPGR protein complex signaling in primary cilia. The cilia in trabecular meshwork (TM) cells has been reported to mediate the intraocular pressure (IOP) sensation. Furthermore, we identified a novel c.A1295G variant in Rho guanine nucleotide exchange factors Gene 40 (ARHGEF40) and a likely pathogenic variant in the RPGR gene, suggesting that they may alter the RhoA activity essential for IOP regulation. Conclusion: Our study supports that low-frequency pathogenic variants in multiple genes and pathways probably affect Primary Open Angle Glaucoma's pathogenesis in the large South Indian family. Furthermore, it requires larger case-controls to perform family-based association tests and to strengthen our analysis.
Genetic characterization of Stargardt clinical cases
Raj, Rajendran Kadarkarai, Manojkumar Kumaran et al. Eye and Vision (2020)
Show Abstract
Background: Stargardt disease 1 (STGD1; MIM 248200) is a monogenic form of autosomal recessive genetic disease caused by mutation in ABCA4. This gene has a major role in hydrolyzing N-retinylidene-phosphatidylethanolamine to all-trans-retinal and phosphatidylethanolamine. The purpose of this study is to identify the frequency of putative disease-causing mutations associated with Stargardt disease in a South Indian population.
Methods: A total of 28 clinically diagnosed Stargardt-like phenotype patients were recruited from south India. Ophthalmic examination of all patients was carefully carried out by a retina specialist based on the stages of fundus imaging and ERG grouping. Genetic analysis of ABCA4 was performed for all patients using Sanger sequencing and clinical exome sequencing.
Results: This study identified disease-causing mutations in ABCA4 in 75% (21/28) of patients, 7% (2/28) exhibited benign variants and 18% (5/28) were negative for the disease-causing mutation.
Conclusion: This is the first study describing the genetic association of ABCA4 disease-causing mutation in South Indian Stargardt 1 patients (STGD1). Our findings highlighted the presence of two novel missense mutations and an (in/del, single base pair deletion & splice variant) in ABCA4. However, genetic heterogeneity in ABCA4 mutants requires a larger sample size to establish a true correlation with clinical phenotype.
Performance assessment of variant calling pipeline using human whole exome sequencing and simulated data
Manojkumar Kumaran et al. BMC Bioinformatics (2019)
Show Abstract
Background: Whole exome sequencing (WES) is a cost-effective method that identifies clinical variants but it demands accurate variant caller tools. Currently available tools have variable accuracy in predicting specific clinical variants. But it may be possible to find the best combination of aligner-variant caller tools for detecting accurate single nucleotide variants (SNVs) and small insertion and deletion (InDels) separately. Moreover, many important aspects of InDel detection are overlooked while comparing the performance of tools, particularly its base pair length.
Results: We assessed the performance of variant calling pipelines using the combinations of four variant callers and five aligners on human NA12878 and simulated exome data. We used high confidence variant calls from Genome in a Bottle (GiaB) consortium for validation, and GRCh37 and GRCh38 as the human reference genome. Based on the performance metrics, both BWA and Novoalign aligners performed better with DeepVariant and SAMtools callers for detecting SNVs, and with DeepVariant and GATK for InDels. Furthermore, we obtained similar results on human NA24385 and NA24631 exome data from GiaB.
Conclusion: In this study, DeepVariant with BWA and Novoalign performed best for detecting accurate SNVs and InDels. The accuracy of variant calling was improved by merging the top performing pipelines. The results of our study provide useful recommendations for analysis of WES data in clinical genomics.
Services

Latest Blog

Bee Eater
The quiet beauty of nature lies in its simplicity, where every flutter of a wing and every whisper of a breeze tells a story of life unfolding..

Drops of Serenity
A single drop holds the universe, reflecting the beauty of nature in its purest form. Just like life, every moment is fleeting yet infinitely profound
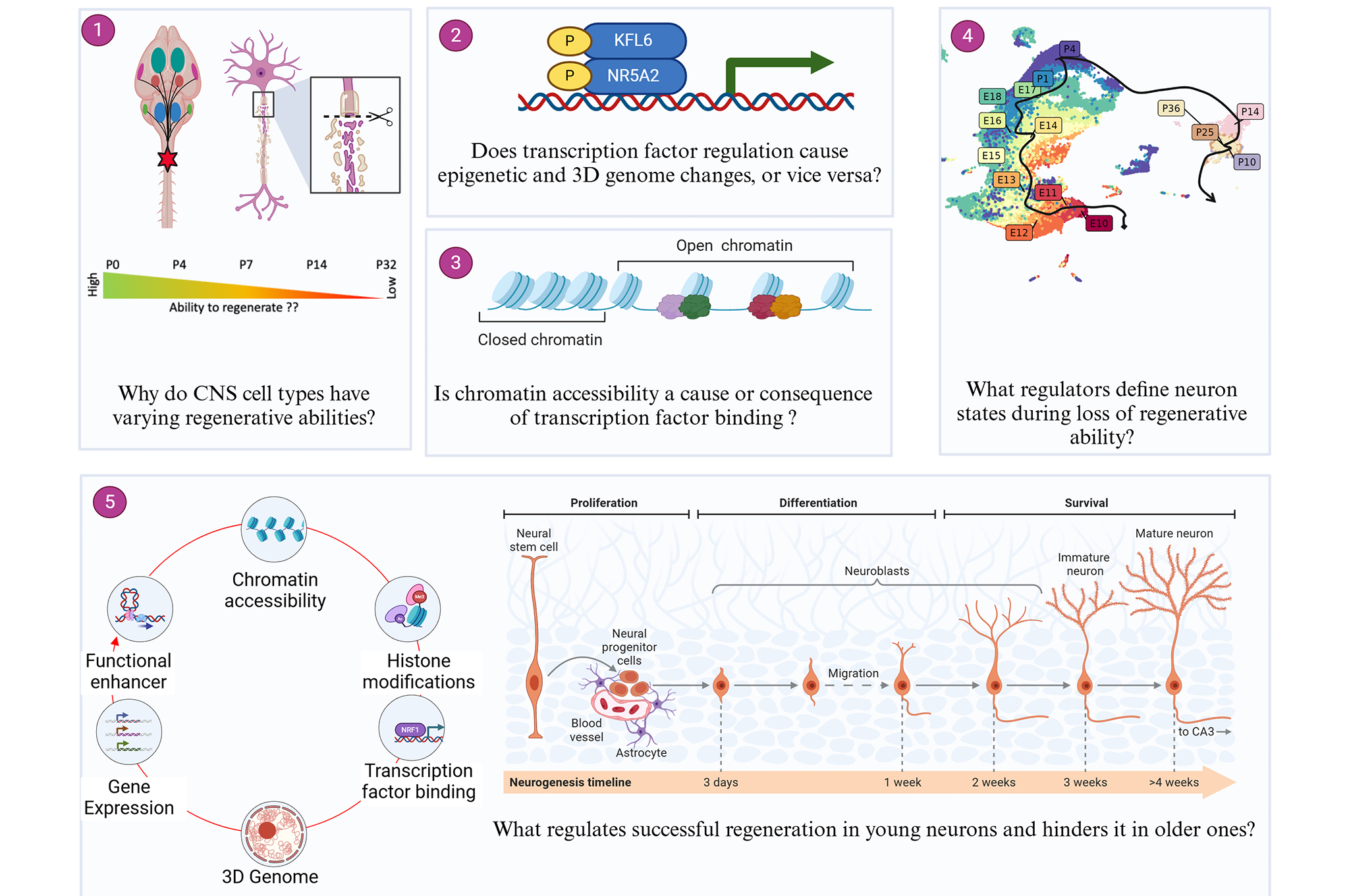
Unraveling Neural Pathways
The brain's complexity is reflected in its genomic blueprint—each gene, a note; each pathway, a melody shaping the orchestra of neural development.
Testimonials

"Manoj is an exceptional bioinformatician. His expertise in genomics and software development is truly inspiring!"
- Dr. Ishwariya Venkatesh

"Working with Manoj has been an absolute pleasure. His ability to automate complex workflows is unmatched."
- Dr. Bharanidharan

"A brilliant researcher with an innovative approach to computational biology. Highly recommended!"
- Vatsal Mehara
Contact Me
Call Me On
+91 7708556754
Office
Centre for Cellular & Molecular Biology, Habsiguda, Uppal, Hyderabad - 500 007
manojkumarbioinfo@gmail.com